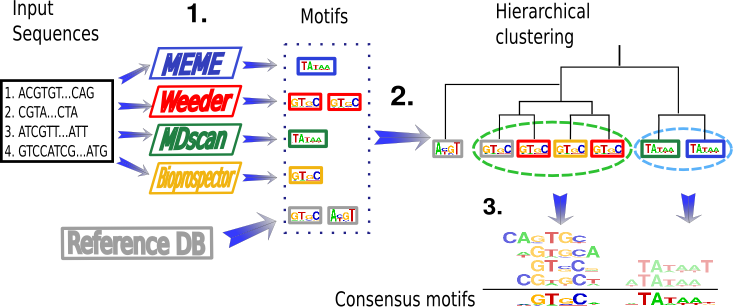
MeMoFinder is a software helping researchers in the task of finding motifs in biological sequences using a consensus approach. This means that instead of developing a new method for finding sequence motifs de novo we have focused on finding the consensus between different available tools and data stored in public motif databases such as JASPAR.
As you can see in the picture above, our method is composed of three steps which are described below.
At the end of this step, the programs' results are gathered together, the results are pre-filtered, so that multiple copies of the same motif reported by any of the programs are removed.
At this stage the motifs from the reference database (either a flavor of JASPAR or user-supplied database, more details) are added to the pool of motifs. This allows to spot the similarities between previously known motifs and the ones reported by de novo methods.
More on motif finding...The matrix is then used as an input to a hierarchical clustering of the motifs using average linkage method (more info... ). The threshold value for clustering can be specified either as an absolute distance, or it can be specified relative to the minimal distance between the motifs in the reference database. The default is to select the relative threshold of 0.5, which results in all reference motifs being in different clusters.
Then motifs in each of the clusters are aligned and a consensus motif for each cluster is computed. This is done using an incremental alignment of the motifs starting from the most informative to the least informative one. The motifs are then truncated so that they do not contain non-informative columns at their ends.
more on clustering...