Supervised learning method for predicting chromatin boundary associated insulator elements
Pawel Bednarz and Bartek Wilczynski
Supplementary figures submitted with the manuscript:
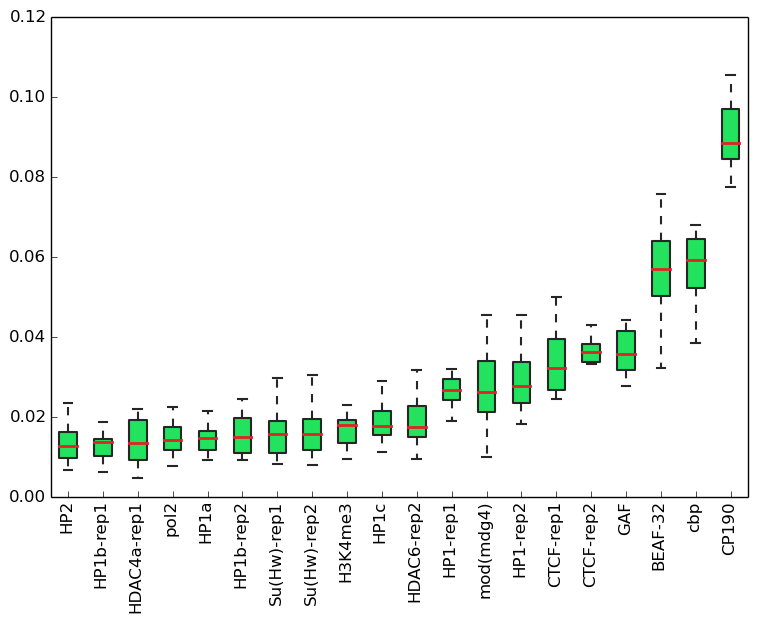
Supplementary Figure 1. The most predictive 20 features used by a random forest classifier trained both on modEncode data and k-mers.
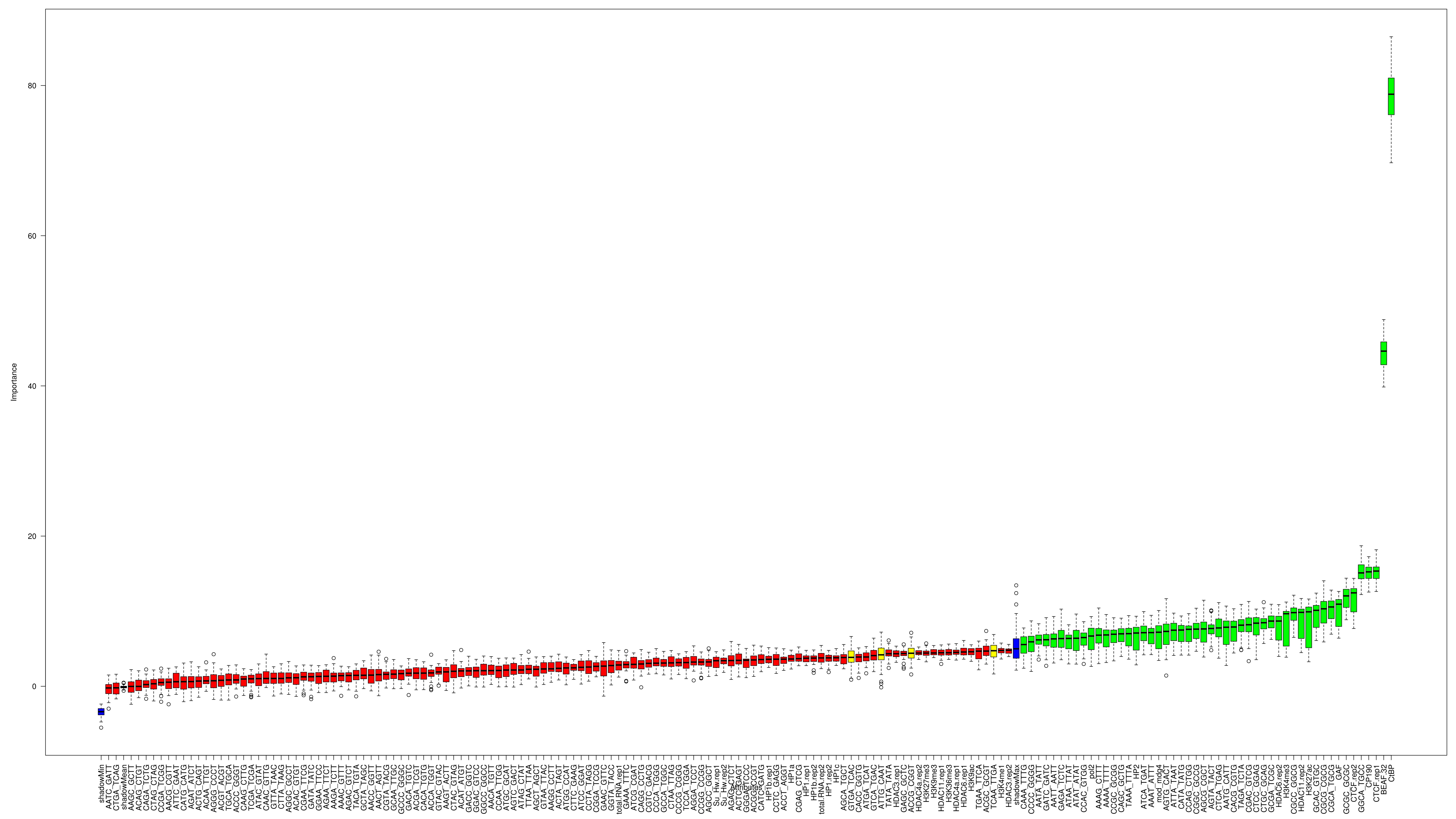
Supplementary Figure 2. The most predictive features used by a random forest classifier trained both on modEncode data and k-mers. Output from Boruta package taking into consideration information content of every variable. We used different window size for averaging modEncode data (2000bp) and different for sequential features (6000bp). Those parameters were chosen because of the good (better than with using the same length for both features) performance of such a classifier.
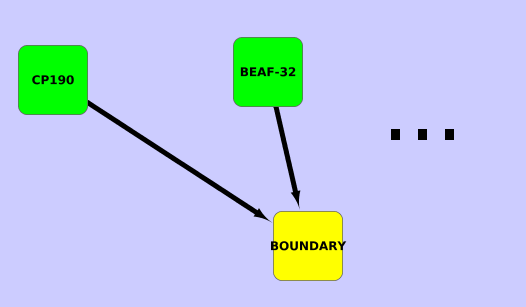
Supplementary Figure 3. Schematic structure of the Bayessian network classifier imposed during learning.
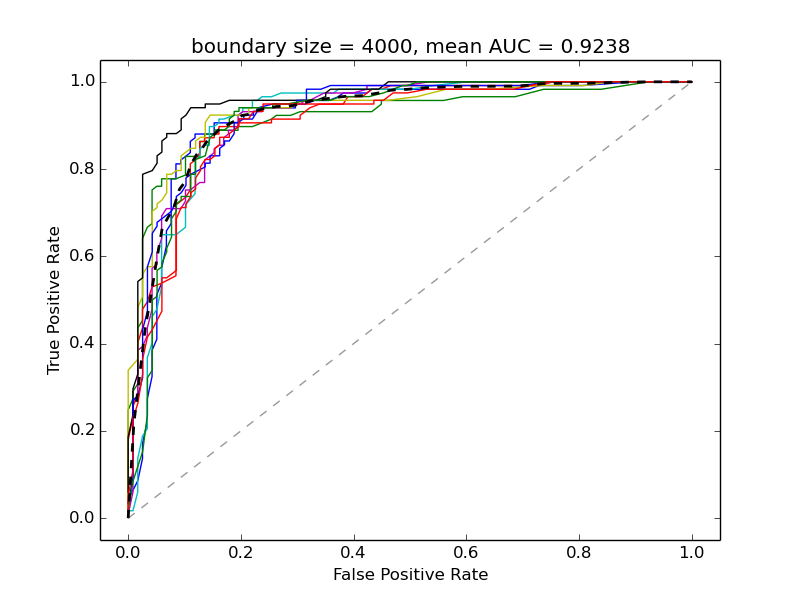
Supplementary Figure 4. Performance of Bayessian network classifier on the boundaries from Sexton et al. trained on modEncode data - 10-fold cross-validation test.
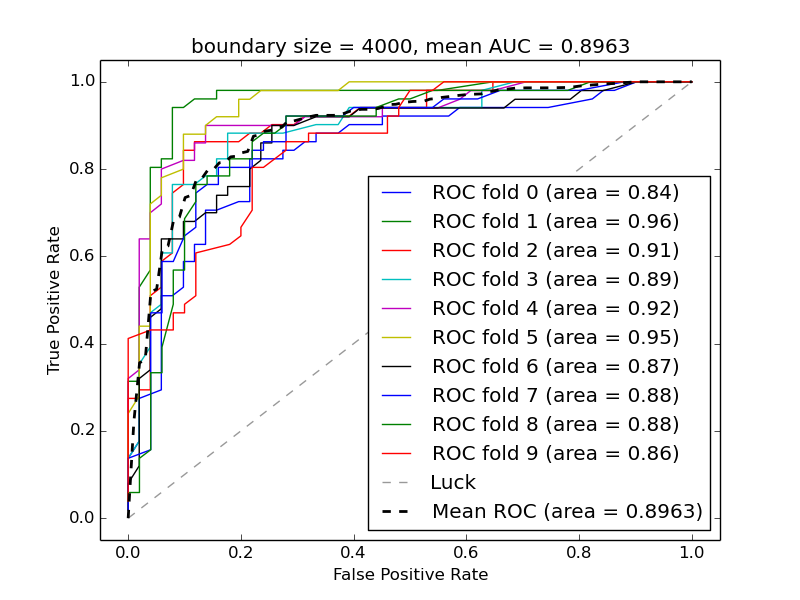
Supplementary Figure 5. Performance of random forest classifier on the boundaries from Sexton et al. trained on modEncode data - 10-fold cross-validation test.
Supplementary Figure 6. Performance of random forest classifier on the boundaries from Sexton et al. without the boundaries from Filion et al. trained on modEncode data - 10-fold cross-validation test.
Supplementary Figure 7. Performance of random forest classifier on the boundaries from Filion et al. without the boundaries from Sexton et al. trained on modEncode data - 10-fold cross-validation test.
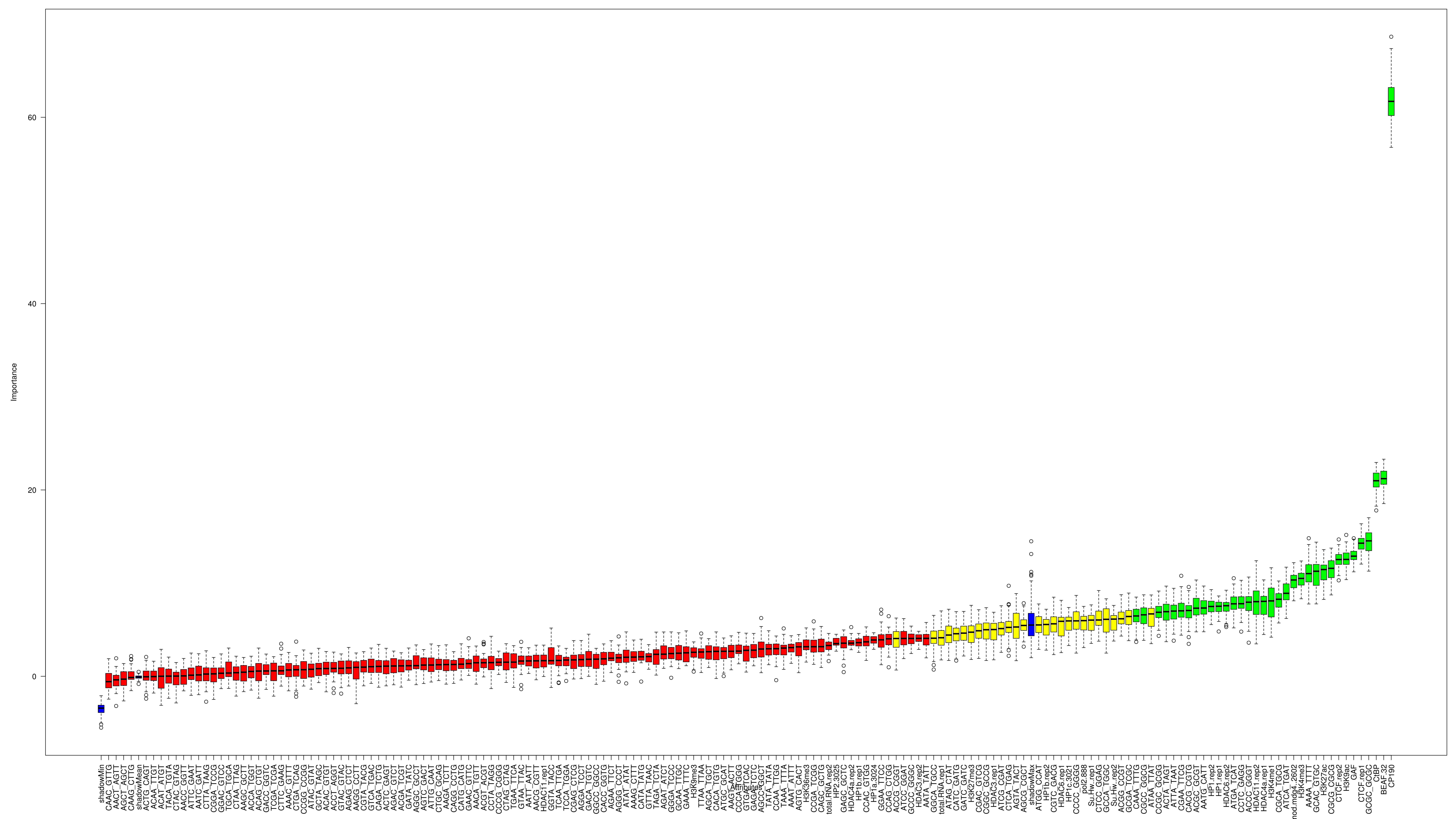
Supplementary Figure 8. The most predictive features used by a random forest classifier trained both on modEncode data and k-mers. Output from Boruta package taking into consideration information content of every variable. Windows of 4000bp were used both for modEncode and sequential features.
Supplementary tables submitted with the manuscript:
Supplementary Table 1. Comparison of performance of the random forest classifier based on k-mers for different values of k parameter.
Supplementary Table 2. The most predictive features used by a random forest classifier trained both on modEncode data and k-mers. Output from Boruta package taking into consideration information content of every variable. We used different window size for averaging modEncode data (2000bp) and different for sequential features (6000bp). Those parameters were chosen because of the good (better than with using the same length for both features) performance of such a classifier.
Supplementary Table 3. Comparison of performance of the Bayessian network classifier trained on modEncode data for different bin sizes.
Supplementary Table 4. Comparison of performance of the random forest classifier trained on modEncode data for different bin sizes.
Supplementary Table 5. Number of domains when we use predictions with probability greater than 0.5. BN - Bayessian networks, RF - random forest, MOD - modEncode training set, SEQ - k-mer-based training set.
Supplementary Table 6. The most predictive features used by a random forest classifier trained both on modEncode data and k-mers. Output from Boruta package taking into consideration information content of every variable. Windows of 4000bp were used both for modEncode and sequential features.
Predictions from the model:
19.12.2014 Due to an error in wig to bed conversion, those (corrupted) bed files were put on the server:
Please do not use them in your analysis.
Binarization of the modEncode data for ChromHMM:
ChromHMM before learning the model needs the data to be binarized in order to reduce complexity of the problem. It means that for every bin of size 4000bp and every modEncode track we had to provide the program with the value 0 or 1. Since ChromHMM's procedure for binarization of the input was developed for dealing with ChIP-Seq data and many of our files were the results from ChIP-Chip experiment, we had to binarize the data ourselves. To do so, we used simililar approach to what the authors of ChromHMM did: we used Poisson model of the background with the parameter λ equal to the average signal of the track over the genome. Then we took one sample of ChIP-Seq data and fitted the threshold for λ so that the method for ChIP-Seq data worked similarly to the original binarization. Then we used the same parameter to binarize all the input data for ChromHMM.
BNFinder invocation (for cross-validation):
bnf-cv -e bnf_training_set.txt -s BDE -c net.cpd -i 10 -l 3 -k 10 -r roc.pdf -f 0.001
Packaged scripts used in the analysis:
scripts.tgz
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}